
IMBRUVICA® FOR
YOUR PEDIATRIC PATIENTS WITH cGVHD
IMBRUVICA® safety and effectiveness have been established for children as young as 1 year of age with previously treated cGVHD2*
Previously treated is defined as failure of one or more lines of systemic therapy.
*This indication is supported by evidence from the iMAGINE study.

cGVHD treatment developed in an oral suspension for children
Pediatric patients under the age of 12 who developed cGVHD and have failed one or more lines of systemic therapy have limited treatment options.1
Data from the iMAGINE trial indicate that IMBRUVICA® produced a clinically meaningful response in pediatric patients with cGVHD.2
Study design: iMAGINE trial2,3
An open-label, multicenter, single-arm, phase 1/2 study consisting of 47 patients aged 1 to <22 years with previously treated moderate or severe cGVHD
Select Inclusion Criteria:
- Platelets ≥30 × 109/L and no transfusion for 7 days
- Absolute neutrophil count ≥1.0 x 109/L and off growth factor support for 7 days
- Total bilirubin ≤1.5x ULN (unless of nonhepatic origin or due to Gilbert's syndrome) or ≤3.0 x ULN if due to cGVHD
- Estimated creatinine clearance ≥30 mL/min
Select Exclusion Criteria:
- Single-organ genitourinary involvement was the only manifestation of cGVHD
Select Patient Demographics:
- Median age of patients was 13 years (range: 1-19)
- Median time since diagnosis was 16.1 months
- Median number of prior cGVHD treatments was 2 (range: 1-12)
- Daily corticosteroid dose (prednisone or prednisone equivalent) at baseline was 0.47 mg/kg/day
Study Design Dosing:
- Patients 12 years and older (n=26): 420 mg taken orally once daily until disease progression.
- Patients 1 to less than 12 years of age (n=21): starting dose of 120 mg/m2, after 14 days of treatment 240 mg/m2 taken orally once daily (up to a dose of 420 mg)
cGVHD=chronic graft versus host disease, ULN=upper limit of normal.
Efficacy in children aged 1 to <22 years with cGVHD who have failed one or more lines of therapy2
Approximately 6 out of 10 patients responded to treatment with IMBRUVICA® based on ORR
The efficacy of IMBRUVICA® was established based on responses evaluated through Week 25 visit where overall response included complete response or partial response according to the 2014 National Institutes of Health Consensus Development Project Response Criteria.
Overall Response Rate (ORR)2*
95% CI: 44%, 74% (N=47)
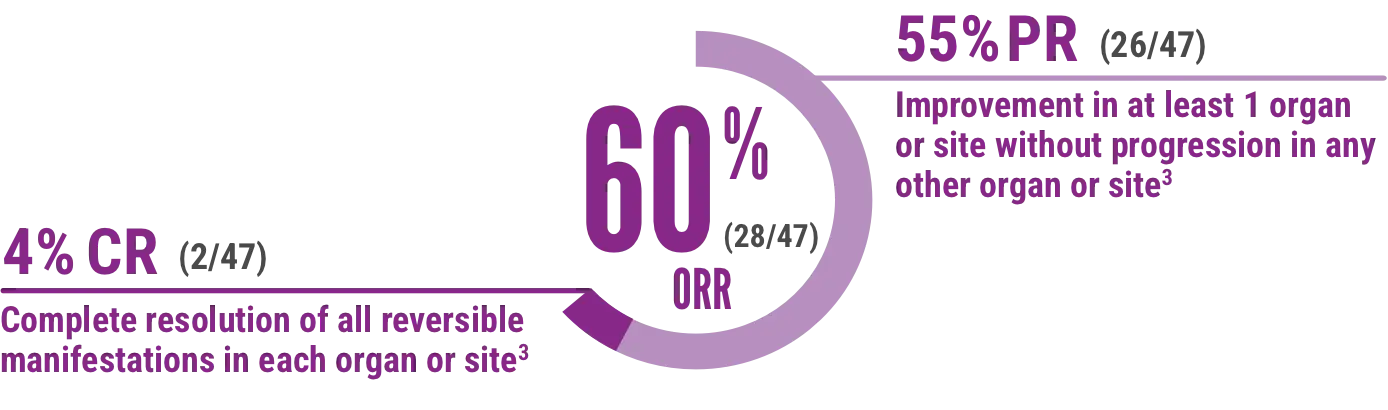
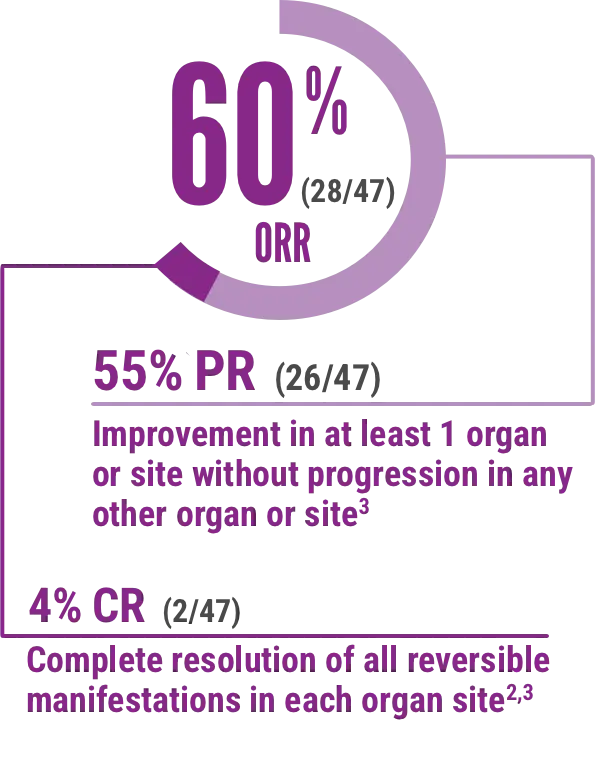
Skin, mouth, liver, upper and lower GI, esophagus, lung, eye, and joint/fascia were the organs/sites considered in evaluating overall response.
CR=complete response, ORR=overall response rate, PR=partial response.
Time to response/duration of response2
Time to first response
Median time to first response was 0.9 month (range: 0.9, 6.1)
The median time from first response to death or new systemic therapies for cGVHD was 14.8 months (95% CI: 4.6, not evaluable).
Median duration of response
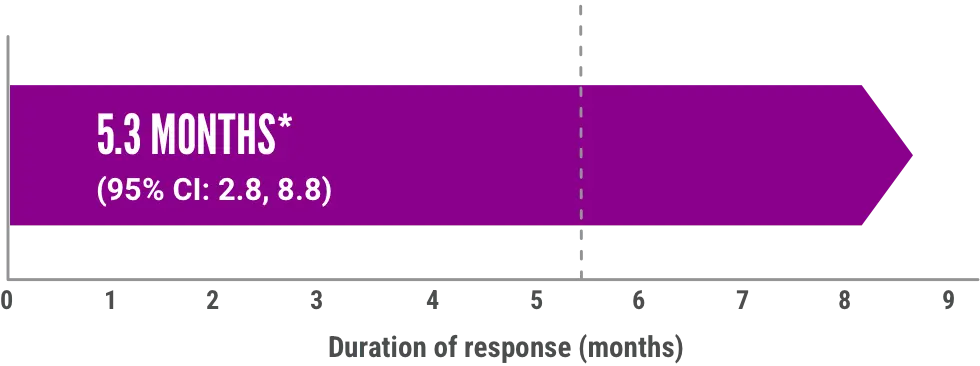
Median duration of response was calculated from first response to progression, death, or new systemic therapy for chronic GVHD.
CI=confidence interval.
Improvement in patient-reported symptom bother2

ORR results were supported by exploratory analyses of patient-reported symptom bother, which showed at least a 7-point decrease in Lee Symptom Scale overall summary score through Week 25 in 50% of patients age 12 years and older†
†The Lee Symptom Scale assesses the severity of cGVHD by directly measuring patient symptom burden based on manifestations of the disease (skin, energy, lung, nutritional status, psychological functioning, eye, and mouth).4
Safety in pediatric patients with cGVHD who have failed one or more lines of therapy2
The iMAGINE trial safety
Median duration of exposure was 7.1 months (range: 0.2, 25.9 months)2
Adverse reactions reported in ≥10% of patients aged 1 to <22 years with cGVHD in iMAGINE2
| Previously Treated (N=47) | ||
|---|---|---|
| Adverse reactions | All Grades (%) | Grade 3 or 4 (%) |
| General disorders and administration site conditions | ||
| Pyrexia | 30 | 11 |
| Musculoskeletal and connective tissue disorders | ||
| Musculoskeletal pain* | 30 | 2 |
| Osteonecrosis | 11 | 9 |
| Gastrointestinal disorders | ||
| Diarrhea | 28 | 2 |
| Abdominal pain* | 23 | 4 |
| Stomatitis* | 23 | 9 |
| Vomiting | 19 | 2 |
| Nausea | 19 | 4 |
| Infections and infestations | ||
| Pneumonia* | 23 | 13 |
| Skin infection* | 17 | 4 |
| Sepsis* | 11 | 9† |
| Nervous system disorders | ||
| Headache | 21 | 2 |
| Skin and subcutaneous tissue disorders | ||
| Rash* | 19 | 2 |
| Pruritus | 13 | 0 |
| Petechiae | 13 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||
| Cough | 19 | 2 |
| Vascular disorders | ||
| Hemorrhage* | 17 | 0 |
| Hypertension* | 11 | 4 |
| Blood and lymphatic system disorders | ||
| Hypokalemia | 15 | 6 |
| Hypogammaglobulinemia* | 11 | 0 |
| Cardiac disorders | ||
| Sinus tachycardia | 11 | 0 |
| Investigations | ||
| Alanine aminotransferase increased | 11 | 2 |
Includes multiple ADR terms.
†Includes 1 event with a fatal outcome.
ADR=adverse drug reaction.
Select hematologic laboratory abnormalities (≥10%) that worsened from baseline in patients who received IMBRUVICA® in the iMAGINE trial2
| IMBRUVICA® (N=47) | ||
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | |
| Hemoglobin decreased | 49 | 13 |
| Platelets decreased | 21 | 4 |
| Neutrophils decreased | 13 | 6 |
Treatment-emergent Grade 4 neutropenia occurred in 3% of patients.
23% of previously treated pediatric and young adult (aged 1 to <22 years) patients who received IMBRUVICA® in the iMAGINE study discontinued treatment due to adverse reactions2
- Adverse reactions that resulted in permanent discontinuation in ≥4% patients included hemorrhage
- Adverse reactions leading to dose reduction occurred in 19% of patients
- Adverse reactions that required dose reduction in at least 2 patients included stomatitis.
The safety and effectiveness of IMBRUVICA® in pediatric patients for indications other than cGVHD have not been established.
IMBRUVICA® is available as a once-daily oral suspension and as capsules or tablets2
See full Prescribing Information for complete dosage and administration details.
Recommended Dosing in Pediatric Patients
Age 1 and <12 years
- 240 mg/m2 administered orally once daily (up to a maximum daily dose of 420 mg) until cGVHD progression, recurrence of an underlying malignancy, or unacceptable toxicity
- When a patient no longer requires therapy for the treatment of cGVHD, IMBRUVICA® should be discontinued considering the medical assessment of the individual patient
Recommended Dosing in Pediatric Patients
- 420 mg administered orally once daily or 6 mL for oral suspension until cGVHD progression, recurrence of an underlying malignancy, or unacceptable toxicity
- IMBRUVICA® can be dosed as a single 420-mg tablet, three 140-mg capsules, or 6 mL for oral suspension
- When a patient no longer requires therapy for the treatment of cGVHD, IMBRUVICA® should be discontinued considering the medical assessment of the individual patient
Recommended dosage based on BSA for patients 1 to <12 years of age using either IMBRUVICA®
capsules/tablets or oral suspension2
| BSA (m2) range | Recommended dose to achieve 240 mg/m2 | |
|---|---|---|
| Volume (mL) of IMBRUVICA® oral suspension (70 mg/mL) | Dose (mg) of IMBRUVICA® tablets/capsules | |
| >0.3-0.4 | 1.2 mL | N/A |
| >0.4-0.5 | 1.5 mL | N/A |
| >0.5-0.6 | 1.9 mL | N/A |
| >0.6-0.7 | 2.2 mL | N/A |
| >0.7-0.8 | 2.6 mL | 210 mg |
| >0.8-0.9 | 2.9 mL | 210 mg |
| >0.9-1 | 3.3 mL | 210 mg |
| >1-1.1 | 3.6 mL | 280 mg |
| >1.1-1.2 | 4 mL | 280 mg |
| >1.2-1.3 | 4.3 mL | 280 mg |
| >1.3-1.4 | 4.6 mL | 350 mg |
| >1.4-1.5 | 5 mL | 350 mg |
| >1.5-1.6 | 5.3 mL | 350 mg |
| >1.6 | 6 mL | 420 mg |
See full Prescribing Information for complete dosage and administration details.
Recommended dosage modifications based on BSA using either IMBRUVICA®
capsules/tablets or oral suspension
| Recommended dose to achieve 160 mg/m2 | Recommended dose to achieve 80 mg/m2 | |||
|---|---|---|---|---|
| BSA (m2) range | Dose (mg) of IMBRUVICA® capsules/tablets to administer | Volume (mL) of IMBRUVICA® oral suspension (70 mg/mL) to administer | Dose (mg) of IMBRUVICA® capsules/tablets to administer | Volume (mL) of IMBRUVICA® oral suspension (70 mg/mL) to administer |
| >0.3-0.4 | - | 0.8 mL | - | 0.4 mL |
| >0.4-0.5 | - | 1 mL | - | 0.5 mL |
| >0.5-0.6 | - | 1.3 mL | - | 0.6 mL |
| >0.6-0.7 | - | 1.5 mL | - | 0.7 mL |
| >0.7-0.8 | 140 mg | 1.7 mL | 70 mg | 0.9 mL |
| >0.8-0.9 | 140 mg | 1.9 mL | 70 mg | 1 mL |
| >0.9-1 | 140 mg | 2.2 mL | 70 mg | 1.1 mL |
| >1-1.1 | 140 mg | 2.4 mL | 70 mg | 1.2 mL |
| >1.1-1.2 | 210 mg | 2.6 mL | - | 1.3 mL |
| >1.2-1.3 | 210 mg | 2.9 mL | - | 1.4 mL |
| >1.3-1.4 | 210 mg | 3.1 mL | - | 1.5 mL |
| >1.4-1.5 | 210 mg | 3.3 mL | 140 mg | 1.7 mL |
| >1.5-1.6 | 280 mg | 3.5 mL | 140 mg | 1.8 mL |
| >1.6 | 280 mg | 4 mL | 140 mg | 2 mL |
BSA=body surface area, CYP3A=cytochrome P450, family 3, subfamily A.
Please see full Prescribing Information for volume of IMBRUVICA® needed to reach the recommended dosage.
- The coadministration of IMBRUVICA® with a strong or moderate CYP3A inhibitor may increase ibrutinib plasma concentrations. Increased ibrutinib concentrations may increase the risk of drug-related toxicity
- After discontinuation of a CYP3A inhibitor, resume previous dose of IMBRUVICA®
- Avoid grapefruit and Seville oranges during IMBRUVICA® treatment, as these contain strong or moderate inhibitors of CYP3A
- Avoid coadministration with strong CYP3A inducers
Administer IMBRUVICA® at approximately the same time each day. Swallow tablets or capsules whole with a glass of water. Do not open, break, or chew the capsules. Do not cut, crush, or chew the tablets. Follow Instructions for Use for further administration details of IMBRUVICA® oral suspension. If a dose of IMBRUVICA® is not taken at the scheduled time, it can be taken as soon as possible on the same day with a return to the normal dosing schedule the following day. Do not take extra doses of IMBRUVICA® to make up for the missed dose.2
Dose Modifications for Adverse Reactions in cGVHD
Dosage modifications for adverse reactions in pediatric patients
If an AR listed below occurs, interrupt IMBRUVICA® therapy at each occurrence of the same AR. Once the AR has improved to Grade 1 or baseline, follow the recommended dosage modifications below.2
Recommended dosage modifications for ARs2
| Adverse reaction*† | Occurrence | Dose modification for patients 12 years or older with cGVHD after recovery Starting Dose = 420 mg | Dose modification for patients 11 to less than 12 years with cGVHD after recovery Starting Dose = 240 mg/m2 |
|---|---|---|---|
| Grade 2 cardiac failure | First | Restart at 280 mg daily‡ | Restart at 160 mg/m2 daily‡ |
| Second | Restart at 140 mg daily‡ | Restart at 80 mg/m2 daily‡ | |
| Third | Discontinue IMBRUVICA® | Discontinue IMBRUVICA® | |
| Grade 3 cardiac arrhythmias | First | Restart at 280 mg daily‡ | Restart at 160 mg/m2 daily‡ |
| Second | Discontinue IMBRUVICA® | Discontinue IMBRUVICA® | |
| Grade 3 or 4 cardiac failure Grade 4 cardiac arrhythmias | First | Discontinue IMBRUVICA® | Discontinue IMBRUVICA® |
| Other Grade 3 or 4 non-hematological toxicities§ | First | Restart at 280 mg daily | Restart at 160 mg/m2 daily‡ |
| Grade 3 or 4 neutropenia with infection or fever | Second | Restart at 140 mg daily | Restart at 80 mg/m2 daily‡ |
| Grade 4 hematological toxicities | Third | Discontinue IMBRUVICA® | Discontinue IMBRUVICA® |
Please see Warnings and Precautions section of the Prescribing Information.
†Grading based on National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) criteria, or International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria for hematologic toxicities in CLL/SLL.
‡Evaluate the benefit-risk before resuming treatment.
§For Grade 4 non-hematologic toxicities, evaluate the benefit-risk before resuming treatment.
Refer to Dosage and Administration (2.2) Table 3 in the Prescribing Information for recommended dosage modifications based on BSA using either IMBRUVICA® capsules/tablets or oral suspension.
AR=adverse reaction, CLL=chronic lymphocytic leukemia, iwCLL= International Workshop on Chronic Lymphocytic Leukemia, NCI-CTCAE=National Cancer Institute-Common Terminology Criteria for Adverse Events, SLL=small lymphocytic lymphoma.
Patients on CYP3A Inhibitors
Patients on CYP3A inhibitors may receive IMBRUVICA®2
In an observational, analytical, retrospective cohort study,
50% of pediatric patients with allogeneic hematopoietic stem cell transplantation used CYP3A inhibitors in the month before transplantation5
Dosage modifications for use with CYP3A inhibitors in pediatric patients with cGVHD2
| Coadministered drug | Recommended IMBRUVICA® dosage | |
|---|---|---|
| Patients ≥12 years of age | Moderate CYP3A inhibitors | 420 mg once daily* |
| 280 mg once daily* | |
| 140 mg once daily* | |
| Other strong CYP3A inhibitors | Avoid concomitant use. If these inhibitors will be used short-term (such as anti- infectives for seven days or less), interrupt IMBRUVICA® | |
| Patients 1 to <12 years of age | Moderate CYP3A inhibitors | 240 mg/m2 once daily* |
| Voriconazole for suspension 9 mg/kg (maximum dose: 350 mg) twice daily | 160 mg/m2 once daily* | |
| Posaconazole at any dosage | 80 mg/m2 once daily* | |
| Other strong CYP3A inhibitors | Avoid concomitant use. If these inhibitors will be used short-term (such as anti- infectives for seven days or less), interrupt IMBRUVICA® |
*Modify dose as recommended.
Refer to Dosage and Administration (2.2) Table 3 in the Prescribing Information for recommended dosage modifications based on BSA using either IMBRUVICA® capsules/tablets or oral suspension.
Please see full Prescribing Information for volume of IMBRUVICA® needed to reach the recommended dosage.
- The coadministration of IMBRUVICA® with a strong or moderate CYP3A inhibitor may increase ibrutinib plasma concentrations. Increased ibrutinib concentrations may increase the risk of drug-related toxicity
- After discontinuation of a CYP3A inhibitor, resume previous dose of IMBRUVICA®
- Avoid grapefruit and Seville oranges during IMBRUVICA® treatment, as these contain strong or moderate inhibitors of CYP3A
- Avoid coadministration with strong CYP3A inducers
Patients With Hepatic Impairment
Dosage modifications for use in hepatic impairment
- The recommended dosage is 140 mg daily for patients 12 years of age and older with total bilirubin level >1.5 to 3x ULN (unless of non-hepatic origin or due to Gilbert’s syndrome)
- The recommended dosage is 80 mg/m2 daily for patients 1 to less than 12 years of age with total bilirubin level >1.5 to 3x ULN (unless of non-hepatic origin or due to Gilbert’s syndrome)
- Avoid the use of IMBRUVICA® in these patients with total bilirubin level > 3x ULN (unless of non-hepatic origin or due to Gilbert’s syndrome)
Oral Suspension and Administration of Oral Suspension
Administration of IMBRUVICA® Oral Suspension for Pediatric Patients ≥1 Year
The following oral suspension administration overview is not meant to replace the Instructions for Use that are supplied with IMBRUVICA®.
Please see the full Instructions for Use for details on preparation and administration information enclosed.
Steps for administering IMBRUVICA® to your pediatric patients
Instruct patients to read the Instructions for Use before administering IMBRUVICA®
oral suspension and each time they get a refill.
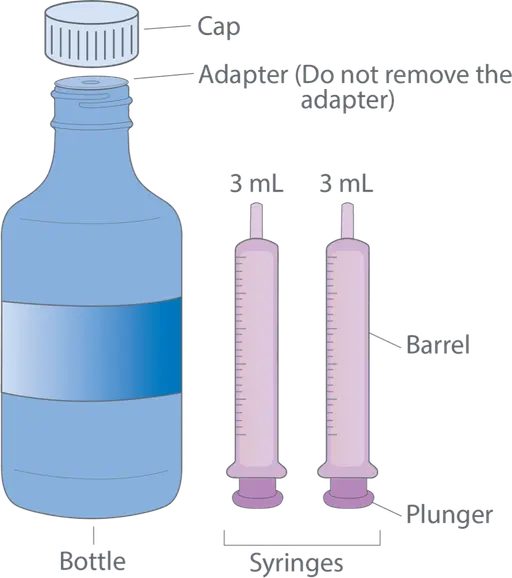
Confirm the dosage of IMBRUVICA®
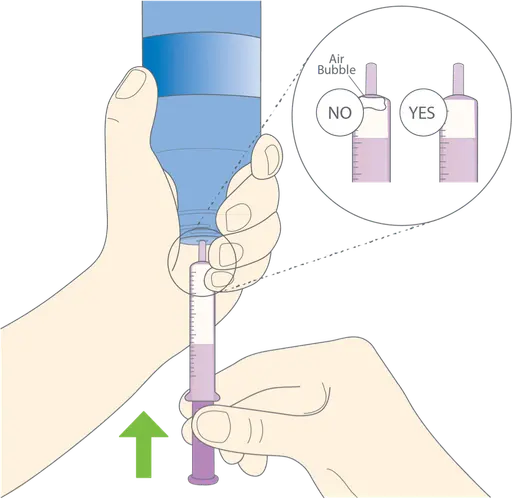
Prepare the dose
Shake bottle well before each use. Press down and twist cap to remove it. Do not remove the adapter. Utilize the provided syringe to withdraw desired dose, removing any air bubbles prior to administration.
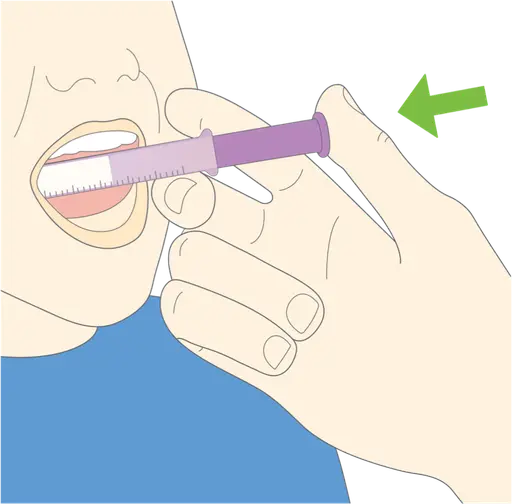
Administer IMBRUVICA®
IMBRUVICA® must be administered as soon as possible after being drawn from the bottle. After swallowing the dose of medicine, make sure the patient drinks water.
How to store IMBRUVICA® oral suspension
How to store IMBRUVICA® capsules
- Store bottles at room temperature 20ºC and 25ºC (68ºF to 77ºF)
- Brief exposure to 15°C to 30°C (59°F to 86°F) permitted (see USP Controlled Room Temperature)
- Retain in original package until dispensing
How to store IMBRUVICA® tablets
- Store bottles at room temperature 20ºC and 25ºC (68ºF to 77ºF)
- Brief exposure to 15°C to 30°C (59°F to 86°F) permitted (see USP Controlled Room Temperature)
- Retain in original package
Click here to learn more about the IMBRUVICA® By Your Side patient support program.
Click here to see NDC numbers for IMBRUVICA® dosing.
Click here to see IMBRUVICA® Diagnosis Codes.